Ischemia is the condition suffered by tissues & organs when deprived of blood flow -- mostly the effects of inadequate nutrient & oxygen. Reperfusion injury refers to the tissue damage inflicted when blood flow is restored after an ischemic period of more than about ten minutes. Ischemia and reperfusion can cause serious brain damage in stroke or cardiac arrest. Cryonics patients frequently experience ischemic & reperfusion injury between the time when the heart stops and cryostorage begins.
In this article I attempt to evaluate the nature & extent of ischemic & reperfusion injury -- primarily focused on the impact for cryonics (although certainly relevant to stroke and cardiac arrest). I also attempt to assess what can be done to minimize such damage. I focus my attention on ischemic/reperfusion injury to the brain. I rely on peer-reviewed journal articles for information. The single most comprehensive article I have found on ischemic and reperfusion injury is "Ischemic Cell Death in Brain Neurons " by Peter Lipton [PHYSIOLOGICAL REVIEWS; 79(4):1431-1568 (1999)]. Most unreferenced factual statements I make are based on Lipton's review.
|
|
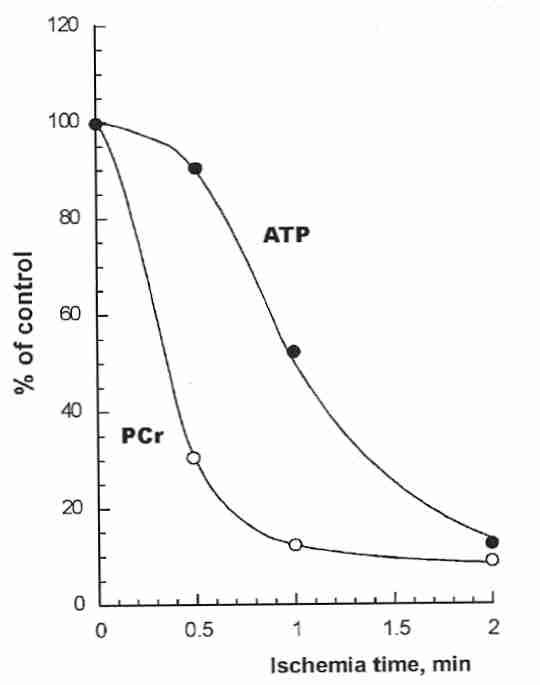
Most of the metabolic energy of neurons is expended on maintaining ion gradients across the cell membrane. A sodium/potassium (Na+/K+) pump keeps extracellular potassium low and extracellular sodium high compared to intracellular concentrations. This pump is driven by the energy stored in ATP (Adenosine TriPhosphate) molecules manufactured in the mitochondria. Most of the energy (ATP) generated in the mitochondria requires oxygen, but in the absence of oxygen some energy can be generated in the cytoplasm outside of the mitochondria by glycolysis, wherein a glucose molecule produces two molecules of ATP and lactate. The liberation of phosphate from ATP is a source of cellular energy that results in ADP (Adenosine DiPhosphate) and hydrogen ion (acid).
In the first minute after stoppage of blood flow to the brain, ATP in neurons is primarily regenerated from ADP by phosphate from PhosphoCreatine (PCr). Within two minutes without blood flow (due to heart stoppage or blood vessel occlusion) neurons lack the energy to power the sodium/potassium pump. Potassium ions rush out of the cell while sodium & chloride ions rush inward as the cell membranes depolarize. The net breakdown of ATP from glycolysis results in ADP, AMP (Adenosine MonoPhosphate), phosphate, lactate and acid accumulation (acidosis). Accumulation of carbon dioxide results in carbonic acid (H2CO3), which further increases acidity. Within two minutes of ischemia, extracellular pH can drop from about 7.3 to about 6.7.
Another ATP-driven pump helps keep extracellular calcium ions (Ca2+)
10,000 times more concentrated than within the cytoplasm. Voltage-gated
ion channels and ion-exchangers in the cell membrane also regulate ion concentrations.
![[NMDA SYNAPSE ILLUSTRATION]](./AmpaNmda.gif)
Depolarization of presynaptic membranes results in release of the neurotransmitter glutamate (glutamic acid). Postsynaptic membranes contain several types of glutamate receptors, notably NMDA & AMPA receptors, which allow calcium ion entry. Postsynaptic membranes contain two voltage-gated calcium channels (L-type & T-type) as well as a sodium/calcium exchanger, but the NMDA channel is particularly adept at allowing large amounts of calcium ion to enter the cell. Excessive glutamate release resulting in excessive Ca+2 entry into cells is the excitotoxicity which initiates the brain ischemic damage seen in stroke and cardiac arrest.
In times of high metabolic demand and adequate availability of oxygen, elevated calcium in mitochondria can increase ATP production by stimulation of three enzymes in the Krebs citric acid cycle: pyruvate dehydrogenase, alpha-ketoglutarate and isocitrate dehydrogenase. But when oxygen is not available in adequate amounts to accept electrons (hydrogen atoms) from NADH, the excess electrons form superoxide from the residual oxygen. Countering NADH production, calcium action on the mitochondrial permeability transition pores increases inner membrane permeability thereby reducing proton potential, causing the matrix to swell and ultimately releasing cytochrome c (an initiator of apoptosis).
High levels of intracellular calcium ion activate proteolytic enzymes (known as calpains) that break down many cell proteins, particularly those in the cytoskeleton of neurons (spectrin, neurofilament and microtubule-associated protein). The fact that Alzheimer's Disease patients have triple the normal levels of calpain in their prefrontal cortex could indicate a role of ischemia as a cause of the disease [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); 90(7):2628-2632 (1993)]. Calcium-activated nuclear endonucleases can cleave chromatin and begin the process of apoptosis ("cell suicide").
Calcium ions also activate phospholipase enzymes which attack cell membrane phospholipids causing the release of arachidonic acid. Inhibitors of the enzymes lipoxygenase & cyclo-oxygenase (which break down arachidonic acid into eicosanoids such as prostaglandin) can reduce cerebral deficits caused by ischemia [CRITICAL REVIEWS OF NEUROBIOLOGY 15(1):61-90 (2003)]. (For more information about phospholipase, eicosanoids, etc., see Essential Fatty Acids in Cell Membranes.)
Most ischemic brain damage is to the lipid portion of cell membranes through lipid peroxidation and phospholipase activity. Cerebral ischemia results in rapid release of fatty acids (especially arachidonic acid) due to phospholipase enzymes. Calcium-dependent cytoplasmic PhospoLipase A2 (cPLA2) is activated by Ca+2 entry into cells after a few minutes of ischemia. cPLA2 preferentially releases oxidized arachidonic acid (which is present in large quantities in neural membranes). Lipoxgenase enzymes form lipid hydroperoxides (ROOH) which can lead to lipid peroxidation by Fenton-like reactions [BIOLOGICAL CHEMISTRY 383:365-374 (2002)]. Arachidonic acid itself has an uncoupling effect on mitochondria in addition to its direct inhibition of mitochondrial respiratory enzymes and promotion of free-radical formation [FREE RADICAL BIOLOGY AND MEDICINE 27(1-2):51-59 (1999)].
Low cell energy and damaged membranes reduce glutamate uptake worsening excitotoxicity. Soon neuron membrane damage is so great that the major mechanism of glutamate release is direct leakage through cell membranes [BRAIN RESEARCH BULLETIN 34(5):457-466 (1994)]. The large (molecular weight 140,000) enzyme Lactate DeHydrogenase (LDH) is soon seen leaking through ischemia-damaged membranes. Blood or tissue levels of LDH have often been used as an indicator of cell damage due to ischemic/reperfusion injury. LDH is very suitable as an assay for cell lysis because it exists in relatively high concentration in all cells, and is stable.
Prompt restarting of circulation following ischemia can prevent tissue damage. Restarting blood flow after more than about ten minutes of ischemia is typically more damaging than the ischemia itself because the ischemia sets the stage for oxygen to generate free-radicals rather than to contribute to cellular energy production [CARDIOVASCULAR RESEARCH; Zweier,JL; 70(2):181-190 (2006)]. In addition to oxygen-generated free radicals, cytokines can be a significant source of reperfusion injury [EMEDICINE; Elzawahry,H; (June 24,2009)]. Two to six hours of ischemia followed by 24 hours of reperfusion more than triples infarct volume [JOURNAL OF CEREBRAL BLOOD FLOW & METABOLISM; Aronowski,J; 17(10):1048-1056 (1997)]. A historical review of oxygen injury due to delayed reperfusion following ischemia can be found in section one of [CARDIOVASCULAR RESEARCH; Zweier,JL; 70(2):181-190 (2006)].
The acidity produced by ischemia greatly
reduces the release of arachidonic acid from cell membranes by phospholipases,
so phospholipase activity During the ischemic period there is an accumulation of lactic acid which lowers cellular
pH. Cells use Na+/H+ exchange to eliminate excess protons, but in
the process accumulate excess Na+ which cannot be exported with the sodium
pump (Na-K-ATPase) due to ATP deficiency. As a consequence cells use the
Na+/Ca+2 exchange, which loads cells with Ca+2.
Upon reperfusion Ca+2 enters the mitochondria, but the
Mitochondrial Permeability Transition
Pore (MPTP) remains closed because the acidity maintains MPTP closure.
Elevation of pH with reperfusion can open the MPTP [BIOCHEMICAL SOCIETY TRANSACTION;
Halestrap,AP; 34(Pt 2):232-237 (2006)]. If the MPTP can close or if ATP can
otherwise be generated cells will die by apoptosis. Without sufficient ATP, MPTP
opening results in necrosis [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS;
Kim,J; 304(3):463-470 (2003)].
NAD(P)H oxidase in reperfusion reacts with newly introduced oxygen to produce
superoxide [STROKE; Kahles,T; 38(11):3000-3006 (2007)]. Superoxide
reacts with iron-sulfur proteins, decreasing their activity and liberating free iron --
which causes hydroxyl radical formation. Nitric oxide in mitochondria reacts with
superoxide three times faster than
SuperOxide Dismutase (SOD).
Superoxide reacts with nitric oxide more efficiently than with any other molecule,
rapidly consuming the nitric oxide to form the potent
free radical
peroxynitrite [JOURNAL OF APPLIED PHYSIOLOGY; Faraci,FM; 100(2):739-743 (2006)].
Peroxynitrite irreversibly inactivates not only SOD, but
complexes I and II of the mitochondrial respiratory chain.
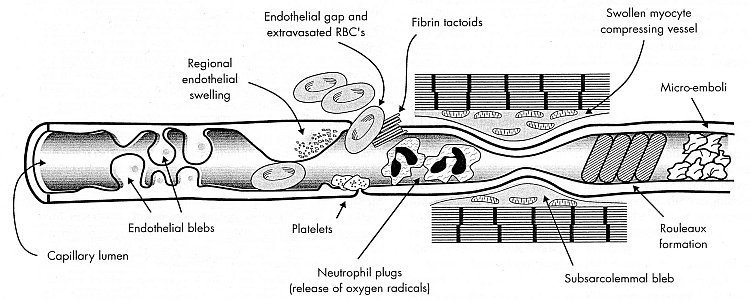
In reperfusion there is considerable membrane damage to endothelial cells as well as
platelets, leucocytes and other cells in the blood stream. Activated neutrophils produce
superoxide, which can be dismutated into hydrogen peroxide. Neutrophil myeloperoxidase enzyme
converts hydrogen peroxide to hypochlorous acid. Hypochlorous acid reacting with superoxide
can produce hydroxyl radicals. Red blood cell aggregation near the exit of capillaries
pushes leukocytes against endothelial cells, thereby increasing leukocyte
adhesion [AMERICAN JOURNAL OF PHYSIOLOGY; Pearson,MJ; 279(4):H1460-H1471 (2000)].
Leukocyte adhesion (and reperfusion damage) is higher in older animals [MICROCIRCULATION;
Ritter,L; 15(4):297-310 (2008)].
Eicosanoids generated by arachidonic acid (especially
leukotrienes) greatly increase the adhesion of leukocytes & platelets to capillary
walls — plugging them up. Superoxide also increases the adhesion of leucocytes to vessel
walls. Leukocyte adhesion is also potentiated by InterCellular Adhesion
Molecule 1 (ICAM−1) protein released from endothelial cell
and leucocyte membranes by cytokines during reperfusion (an effect attenuated by
hypothermia) [STROKE; Ishikawa,M; 30(8):1679-1686 (1999)].
Eicosanoids (leukotrienes & prostaglandins) and associated
oxygen free-radicals make capillary walls more "leaky", causing
edema
which narrows the channels. ATP depletion significantly reduces the ability of
erythrocytes to
deform [THE JOURNAL OF CLINICAL INVESTIGATION; Weed,RI; 48(5):795-809 (1969)].
These effects quickly become pronounced enough in
reperfusion to block capillaries entirely — the no-reflow phenomenon.
Experimental middle cerebral artery occlusion has shown blood flow reduction to 71% of
control after a one hour occlusion and reduction to 22% of control after a four hour
occlusion [BRAIN RESEARCH; Dawson,DA; 749:200-208 (1997)]. The cerebral cortex, the
part of the brain in which
consciousness is presumed to reside, is fortunately less vulnerable to no-reflow than
other areas of the brain. More than 50% of blood vessels have been shown to be
occluded in the thalamus and basal ganglia after 30 minutes of ischemia, but less
than 15% of vessels in the cerebral cortex are
occluded [STROKE; Fischer,EG; 3(5):538-542 (1972)].
But no-reflow can occur even without blood cells. Free-radical
and other membrane damage can loosen or dislodge atherosclerotic plaque causing
emboli upon reperfusion.
Nitric oxide normally functions to not only reduce platelet aggregation &
leukocyte adhesion to the endothelium, but to promote vascular smooth muscle relaxation
and reduce endothelial cell cytokine production. Nitric oxide concentrates in lipophilic
cellular regions with a partition coefficient of 8:1, and can inhibit lipid peroxidation a
thousand times more potently than alpha-tocopherol [JOURNAL OF BIOLOGICAL CHEMISTRY; Rubbo,H; 269(42):26066-26075 (1994)].
Nitric oxide potentiates transcription of
phase 2 detoxification enzymes
(including antioxidant
enzymes) [JOURNAL OF BIOLOGICAL CHEMISTRY;Dhakshinamoorthy,S; 279(19):20096-20107
(2004)]. Nitric oxide inhibits the expression
of pro-inflammatory genes by transcription factor NF-kappaB [TRANSPLANTATION
PROCEEDINGS 30:4239-4243 (1998)]. NF-kappaB activates the cytokine TNF−α to increase
expression of cell adhesion molecules. Nitric oxide inhibits apoptosis by inhibition of
caspase-3 enzyme [JOURNAL OF BIOLOGICAL CHEMISTRY; Rossig,L; 274(11):6823-6826 (1999)].
But these beneficial actions of nitric oxide are seen in the absence of ischemia/reperfusion
— which converts nitric oxide into a toxin.
Elevated blood levels of the pro-inflammatory cytokine TNF−α induces apoptosis [BIOCHEMICAL
AND BIOPHYSICAL RESEARCH COMMUNICATIONS; Bajaj,G; 345(4):1558-1564 (2006)]. In inflammatory
conditions, such as occurs in reperfusion, inducible nitric oxide
synthetase can increase nitric oxide concentration to thousands of times normal
levels [FREE RADICAL BIOLOGY & MEDICINE; Brown,GC; 33(11):1440-1450 (2002)].
During reperfusion,
abnormally high amounts of superoxide converts almost all available nitric oxide to
perxoynitrite — regarded as the agent causing most of the damage to brain capillary
endothelial cells [NEUROSURGERY 43(3):577-584 (1998)]. In one study, inhibition
of reactive peroxynitrite resulting from reperfusion after 30 minutes of warm
ischemia doubled recovery of contractile
function [JOURNAL OF BIOLOGICAL CHEMISTRY; Wang,P; 271(46):29223-29230
(1996)]. Damage to the endothelium
not only increases edema (tissue swelling due to "leakiness"), but causes
endothelial protrusions ("blebs") which can block capillaries.
Ischemia in tissues and blood vessels results in large amounts of ATP being
broken-down to xanthine.
Reperfusion allows the endothelial enzyme xanthine oxidase to convert xanthine plus
oxygen to superoxide & uric acid. Liberated iron & zinc ions further increase
free radical damage. In contrast to the vasculature, mitochondria in tissues
rather than xanthine oxidase are the primary source of oxygen free radicals during
reperfusion
injury [JOURNAL OF CLINICAL INVESTIGATION; 91(2):456-464 (1993)]. But
xanthine oxidase-produced superoxide (and resulting peroxynitrite) damage to endothelial cells
may be the primary mode of reperfusion damage, with far less damage to parenchymal cells, and
far less injury due to neutrophils [SURGERY; Ratych,RE; 102(2):122-131 (1987)].
There is a linear correlation between the amount of reperfusion injury and
disruption of the
blood-brain
barrier (BBB). Water flow into the brain due to BBB disruption can lead to
edema. Further
BBB damage can transform an ischemic stroke into a hemorrhagic stroke.
Proteases (enzymes that degrade proteins) are released in
ischemia [STROKE; Fukuda,S; 35(4):998-1004 (2004)].
Matrix MetalloProteinase−13 (MMP−13, a collagenase)
originating from an unknown source early in ischemia exerts a corrosive effect on
the blood-brain
barrier [STROKE; Rosell,A; 36(7):1415-1420 (2005)], but unlike other MMPs
does not continue to increase in quantity with
time [STROKE; Horstmann,S; 34(9):2165-2170 (2003)]. Leukocytes (neutrophils,
probably) activated by ischemic inflammation release increasing amounts of
MMP−9 (gelatinase−B) which also degrades the blood-brain
barrier [AMERICAN JOURNAL OF PHYSIOLOGY; Gidday,JM; 289(2):H558-H568 (2005)].
Prior to activation of ischemic inflammatory processes, however, reperfusion can
activate gelatinase A (MMP−2), which increases capillary
permeability and hemorrhage, in addition to opening the blood-brain
barrier [STROKE; Rosenberg,GA; 29(10):2189-2195 (1998)].
(For more on "No-reflow", see Reducing "No-reflow". For more on ischemia/reperfusion damage
to the blood-brain barrier leading to edema, see
Edema in Cryonics.)
Can drugs help prevent ischemic damage in cryonics patients? A study of
the literature on stroke therapy is instructive.
One might think that drugs blocking calcium ion entry via NMDA receptors
would be beneficial for stroke, but clinical trials with these substances have
been a failure. Although animal studies show NMDA-blockers to be effective
for the first 4 minutes, after 8 minutes intracellular levels of calcium ion are
the same whether NMDA-blockers are used or not. L-channel blockers (like
nimodipine) make no difference.
There are plausible reasons why NMDA-blockers — even when combined
with L-channel blockers — are of limited usefulness in preventing calcium
entry into ischemic cells. Low levels of ATP mean reduced capacity of the
calcium-ATP pump to keep calcium out of the cell. High cytoplasmic sodium
means high activity of the membrane sodium/calcium exchangers —
particularly those on mitochondrial membranes, which further depletes ATP.
Blockage of L-channels leaves T-channels unblocked. And phospholipase
breakdown products help to release large amounts of calcium ion which has
been bound to the endoplasmic reticulum.
DiHydroPyridine (DHP) derivatives, such as nimodipine, block L-type calcium
channels. But the main benefit of DHPs in ischemia seems to be through arteriole
dilatation rather than neuron calcium-channel blocking. Pre-treatment of
dogs with nimodipine prior to ten minutes of ischemia led to an 80% normal
recovery rate, as compared with an 86% death rate in untreated controls.
Treatment 2 minutes post-ischemia had a negligible effect. [PHARMACOLOGY
OF CEREBRAL ISCHEMIA, Joseph Krieglstein, Editor, p.65-73 (1988)].
Animal studies have shown benefit from antioxidants such as
Vitamin E [BRAIN
RESEARCH 510:335-338 (1990)],
melatonin & nifedipine [JOURNAL
OF PINEAL RESEARCH 33:87-94 (2002)], resveratrol [BRAIN RESEARCH 958:439-447 (2002)],
deprenyl [JOURNAL
OF NEURAL TRANSMISSION 107:779-786 (2000)], and
PBN [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); 92(11):5057-5061
(1995)]. Local anaesthetics have the potential to reduce ischemic damage to brain tissue
by blocking sodium (Na+) channels — reducing electrical activity &
metabolic rate beyond what can be achieved with barbiturates [PHARMACOLOGICAL REVIEWS
48(1):21-67 (1996)]. But all these agents have
failed to pass clinical trials and be accepted as therapeutic agents. Currently,
the only accepted drugs used for stroke therapy are thrombolytics,
anticoagulants and antiplatelet drugs.
Degradation of the fibrin in blood clots by the protease (protein-digesting enzyme)
plasmin requires conversion of plasminogen to plasmin by
tissue
plasminogen activator (tPA). Administering tPA is useful for breaking-up
blood clots, but only when given within 3 hours of the onset of stroke. When
given within 90 minutes of stroke, tPA can more than double the 3-month survival of stroke
patients [NEUROLOGY 55(11):1649-1655 (2000)]. Because of the risk of reperfusion
injury or hemorrhage, thrombolytics are also avoided on patients with severe hypertension,
of advanced age or with evidence of cerebral edema. Mannitol has been used to reduce
cerebral edema, but not in stroke [PROGRESS IN CARDIOVASCULAR DISEASES
42(3):209-216 (1999)].
Because the plasmin produced by tPA is a non-specific protease it not only
dissolves clots, it contributes to vascular degradation and opening of the
blood-brain
barrier by Matrix MetalloProteinases
(MMPs) [STROKE; Pfefferkorn,T; 34(8):2025-2030 (2003)], and can thereby
worsen damage from reperfusion injury if given in delayed reperfusion. Treatment with
tPA is generally deemed to do more harm than good if given more than 3 hours
after a
stroke [STROKE; Clark,WM; 31(4):811-816 (2000)] and at any time if
the stroke affects a large area of the
brain [JOURNAL OF NEUROLOGY, NEUROSURGERY, AND PSYCHIATRY;
65(1):1-9 (1998)]. Tetracyclines,
particularly minocycline, have been shown to not only reduce ischemia-associated
inflammation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Yrjanheikki,J;
96(23):13496-13500 (1999)], but to inhibit
MMPs [BMC NEUROSCIENCE; Machado,LS; 7:56 (2006)]. Inhibition of the
inflammatory cytokine IL−1β can significantly reduce stroke infarct
volume [JOURNAL OF CEREBRAL BLOOD FLOW & METABOLISM; 65(1):1-9 (1998)].
In many cases strokes can resolve spontaneously within a matter of days,
but the cause of this "recanalization" is
uncertain [STROKE; Molina,CA; 32(5):1079-1084 (2001) and
ARCHIVES OF NEUROLOGY; Kassem-Moussa,H; 59(12):1870-1873 (2002)].
Prior to tPA, streptokinase
& urokinase were the most efficacious thrombolytics. The anticoagulant
heparin is given in the hospital and warfarin is used for long-term maintenance.
It is common practice for
low molecular weight heparins
to be given in hospitals as prophylaxis against
deep vein thrombosis, as for chronically bedridden cancer
patients [JOURNAL OF ONCOLOGY PHARMACY PRACTICE; Nishioka,J;
13(2):85-97 (2007)]. Aspirin may be used as an antiplatelet agent.
These therapies cannot be used
for hemorrhagic stroke because they worsen that condition.
For cryonics purposes
streptokinase is the thrombolytic of choice because
a dose of tPA costs thousands of dollars, whereas streptokinase costs a
few hundred dollars. Steptokinase can be ordered from
Sigma-Aldrich
(CAS Number 9002-01-1) or other suppliers of medicine.
Animals that hibernate or estivate are able to avoid or significantly reduce ischemic
damage by reducing their metabolism. The protective mechanisms used by estivators &
hibernators can provide insight into the nature of ischemic damage and possibly into
means to prevent such damage.
Estivation is a state of aerobic hypometabolism that protects animals from dry
(often hot) conditions. Alterations in metabolism associated with estivation include
water retention, greatly reduced protein synthesis, reduced ion pumping, urea accumulation,
and reliance on lipid oxidation (rather than glycolysis) for energy — associated with
greatly reduced cytochrome c oxidase activity in mitochondria [COMPARATIVE
BIOCHEMISTRY AND PHYSIOLOGY PART A; Storey,KB; 133:733-754 (2002)]. Cardiolipin
is a phospholipid that is synthesized exclusively in the mitochondria and is required for
maximal electron transport activity. Cardiolipin content of mitochondria from estivating
snails is reduced 80%, associated with a similar reduction of cytochrome c
oxidase activity [AMERICAN JOURNAL OF PHYSIOLOGY; Stuart,JA; 275(6Pt2):R1977-R1982 (1998)].
Toxic ammonia accumulation is prevented by increased urea synthesis, despite the fact that
this requires energy [JOURNAL OF EXPERIMENTAL BIOLOGY; Chew,SF; 207:777-786 (2004)].
In hibernating arctic squirrels the leucocyte count drops up to 100-fold, which protects
against the "no-reflow" leukocyte adhesion phenomenon associated with disrupted or
greatly reduced blood flow [FREE RADICAL BIOLOGY & MEDICINE; Drew,KL; 31(5):563-573
(2001)]. Heart rate may be reduced 100-fold, metabolic rate reduced to less than 5% of
normal and body temperature can drop to near 0ºC for small mammalian hibernators.
(In non-hibernating mammals temperatures of 10ºC to 20ºC will stop the heart.)
Passive efflux of K+ and passive influx of Na+ is reduced.
Ca2+ sequestering is enhanced. Reliance on lipid hydrolysis as the primary
source of energy results in ketones bodies which may protect the brain against hypoxia
damage [PHYSIOLOGICAL REVIEWS; Carey,HV; 83:1153-1181 (2003)]. Changes in expression
of the transcription factor protein Hypoxia Inducible Factor (HIF-1) may induce
expression of hibernation-regulatory genes [BIOCHEMICA BIOPHYSICA ACT; Morin,P;
1729(1):32-40 (2005)].
Cold ischemia, such as is experienced by some hibernators and by transplantable
organs being preserved at low temperatures, has unique characteristics distinguishing it
from warm ischemia. Unlike cold ischemia, warm ischemia inhibits nitric oxide synthase and
results in production of eicosanoid vasoconstrictors during reperfusion [TRANSPLANTATION
PROCEEDINGS; Hansen,TN; 32:15-18 (2000)]. Although cold temperature can reduce ischemia, it
can introduce new forms of damage, such as chilling injury. Unlike warm ischemia, cold ischemia is also to
associated with an increase in chelatable iron which opens the
Mitochondrial Permeability Transition
Pore (MPTP), usually leading to apoptosis or (more often) necrosis.
This phenomenon has been demonstrated in the absence of increased superoxide or
hydrogen peroxide for liver endothelial cells, particularly, but also for other
tissues [JOURNAL OF HEPATOLOGY; Rauen,U; 40(4):607-615 (2004)].
Some neuroprotective agents that have not passed clinical trials for stroke
therapy have shown to be of demonstrable benefit in preservation of organs
for transplant. Explanations for the benefits of the ingredients used in the
organ-preservation solution Viaspan® (developed as UW Solution
— University of Wisconsin) can be found on the
Viaspan® website or in
[TRANSPLANTATION; Belzer,FO; 45(4):673-676 (1988)].
Allopurinol inhibits xanthine oxidase, blocking the conversion of
xanthine & oxygen to superoxide & uric acid. Glutathione is used as an
antioxidant with membrane-stabilizing properties. Hypothermia may actually increase
permeability of cells to glutathione [CRYOBIOLOGY; Vreugdenhil,PK; 28:143-149 (1991)].
Dexamethasone can also stabilize membranes, but its actual benefit in Viaspan
is dubious. Magnesium seems to counteract some of the effects of
intracelluar calcium and the sulfate ion resists cell swelling because it
is relatively impermeable to cell membranes.
ATP (Adenosine TriPhosphate) rapidly degrades to adenosine, inosine and hypoxanthine,
all of which easily cross cell membranes and can be lost by diffusion. To counteract loss of
ATP, adenosine
(adenine connected to ribose) is added to provide more substrate for ATP synthesis.
Adenosine also reduces adherence of neutrophils to endothelium as well as inhibiting
neutrophil production of reactive
oxygen species [AMERICAN JOURNAL OF PHYSIOLOGY 257(2 Pt 2):H1334-H1339 (1989)].
Monobasic potassium phosphate also supplies substrate for ATP synthesis while
opposing acidification (from anaerobic glycolysis & lactic acid production) and
potassium-leakage. Potassium hydroxide also maintains a high pH while
opposing potassium-leak.
HydroxyEthyl Starch (HES) is added to UW Solution for oncotic support, ie,
to prevent edema in the interstitial space by keeping more fluid in the blood vessels
(a role normally played by blood albumin). HES reduces leucocyte adhesion to blood vessels during
reperfusion [STROKE; Kaplan,SS; 31(9):2218-2223 (2000)].
Although HES is of most value for perfusion, it has been shown to be of benefit
for improved cold storage of organs [TRANSPLANTATION; Southard,JH; 49(2):251-257 (1990)].
Because HES is difficult to obtain and can cause microcirculatory disturbances,
PolyEthylene Glycol (PEG) has been used as a replacement for HES with good
results [THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS; Faure,J;
302(3):861-870 (2002) and JOURNAL OF GASTROENTEROLOGY AND HEPATOLOGY; Franco-Gou,R;
22(7):1120-1126 (2007)].
Dextran−40
(molecular weight 40 kilodaltons) inhibits cell clumping and can replace HES as a less
viscous oncotic agent which is readily excreted by the kidneys.
HEPES is a
zwitterion
buffer which is large enough (238 daltons) to provide extracellular
osmotic support. The ionization constant of water decreases (pKw increases)
as temperature decreases, which means that the pH will rise with temperature decline.
The pK of phosphate and bicarbonate buffers do not change much with
temperature, but the pK of HEPES buffer rises with falling temperature,
thereby compensating for the rising pK of water. Thus HEPES is a better
buffer than phosphate or bicarbonate for maintaining protein (enzyme) structure
and function in hypothermia [CRYOBIOLOGY; Baicu,SC; 45(1):33-48 (2002)].
Lactobionate and raffinose are large molecules added for osmotic support
and to prevent cell swelling which would result from reduced sodium pump activity.
Lactobionate is a strong chelator of calcium and iron ions. Calcium can worsten
ischemic damage, but a calcium-free solution will increase membrane permeability to
calcium, thereby worsening the effects of subsequent calcium exposure (the
"calcium paradox"). Only very small amounts of calcium are necessary to
prevent the calcium paradox [CIRCULATION; Marban,E; 80(6 Suppl):IV17-22 (1989)].
The Penicillin in UW Solution can prevent bacterial growth.
Insulin can increase glucose uptake by cells, but glucose is omitted from UW Solution
in order to reduce cellular acidosis (lactic acid production by glycolysis).
Viaspan® (UW solution) has been reported to be contaminated with iron and to lose
glutathione prior to use [TRANSPLANTATION; Salahadeen,AK; 70(10):1424-1431 (2000)].
Viaspan does not reduce the extreme loss of mitochondrial and cellular calcium by unknown
causes associated with hypothermia [TRANSPLANTATION; Kim,J; 65(3):369-375 (1998)].
A number of new additives have been proposed for organ transplantation solutions to
prevent cold ischemic injury. Dopamine, for example, reduces cold-ischemic
oxidation [AMERICAN JOURNAL OF TRANSPLANTATION; Yard,B; 4:22-30 (2004)]. But free-radical
damage associated with cold ischemia is evidently primarily due to a hypothermic
release of iron. It would therefore be far more effective to eliminate the source of free
radicals by the use of an iron chelator [JOURNAL OF INVESTIGATIVE MEDICINE; Rauen,U;
52(5):299-309 (2004)]. Deferoxamine has been used for this purpose, but
a novel tetraazaannulene derivative (TAA−1) has been shown to completely
inhibit cold-induced injury resulting from chelatable iron release [FREE RADICAL
BIOLOGY & MEDICINE; Rauen,U; 37(9):1369-1383 (2004)].
Glycine reduces hypoxic injury by reducing ion fluxes through the
plasma membrane of Na+ &
Ca2+ [JOURNAL OF HEPATOLOGY; Frank,A; 32:58-66 (2000)]. The ability
of glycine to affect Cl- flux is not relevant for this protective effect.
Glutamine inhibits proteolysis and can activate heat-shock protein, while the addition
of other amino acids can have a nutritional benefit [LIVER TRANSPLANTATION;
Bessems,M; 11(11):1379-1388 (2005)]. Carbon monoxide releasing compounds have a
protective vasodilatory effect and increases mitochondrial respiration after
cold ischemia and reperfusion [KIDNEY INTERNATIONAL; Sandouka,A; 69(2):239-247 (2006)].
Although Viaspan® was treated as a univeral hypothermic preservation solution
for nearly a decade, in the mid-1990s "intracellular-type" solutions with
high potassium such as Hypothermosol® proved to be superior for preserving
hearts & lungs, as well as other cells and tissues.
(For more on organ preservation solutions, see
Blood Washout & Replacement
and Reducing Ischemic Damage by Cooling.)
Nanotechnology may be able to repair freezing damage because brain structure
remains, though in a scrambled form. Unlike freezing damage, warm ischemia eventually
leads to dissolution of brain tissue into a structureless soup.
On the other hand, claims that a few hours of warm ischemia means
certain loss of personal identity cannot be supported. Even after two
hours of warm ischemia (without reperfusion) lysosomal membranes
in cat brain cells remain intact [VIRCHOWS ARCHIV B 25:207-220
(1977)]. Monkey brains subjected to an hour of warm ischemia and
protected from reperfusion injury show short-term recovery [JOURNAL
OF CEREBRAL BLOOD FLOW AND METABOLISM 6(1):15-33 (1986)].
Post-mortem mouse brains subjected to 6 hours of room temperature and
another 18 hours at 4ºC show half the neurons to be morphologically
intact [VIRCHOWS ARCHIV B 63:331-334 (1993)]. Neurons in brain tissue extracted
from humans postmortem for 3 to 6 hours have been shown to recover
oxidative metabolism and axon transport after suitable in-vitro treatment [THE
LANCET 351:499-500 (1998)]. Adult rats subjected to
cerebral ischemia showed no signs of neuron necrosis for 2 hours, and only by 6 hours
did more than 15% of neurons appear
necrotic [STROKE; 26(4):636-643 (1995)].
Similar results have been seen for humans [ANNALS OF
NEUROLOGY; 2:206-210 (1977)].
The CA1 pyramidal neurons of the hippocampus are often regarded to
be the most sensitive to ischemic injury of all neurons. Following 30 minutes
of ischemia and subsequent reperfusion, the CA1 neurons invariably die after
2 or 3 days whereas the reputedly resistant striatal neurons begin to die
after several hours [ANNALS OF NEUROLOGY 11:491-498 (1982)]. In either
case, a cryonics patient should be in a low-temperature condition well before
that time.
Cell death by apoptosis ( "cell suicide ") is a controlled process
by which cells die in a slow and orderly manner so as to be removed by macrophages.
Necrosis, by contrast, is more rapid — leading to cell membrane rupture, spilling
of cell contents and inflammation. Apoptosis requires DNA transcription, new protein
synthesis — a process requiring many hours, if not days.
The rapidity & form of cell death has been shown to be a function of the degree
of ATP depletion. Mouse kidney cells in which ATP levels were 15% or less than normal
(less than control) died by necrosis over a period between 2 and 4 hours. Cells
with ATP levels 25% of normal remained viable for at least 6 hours, but had all
experienced apoptotic death by
48 hours [AMERICAN JOURNAL OF PHYSIOLOGY; 274(2 Pt 2):F315-F327
(1998)].
Apoptosis is probably no ultimate hazard for cryonics patients
who deanimate without pre-mortem ischemic damage and who receive
prompt cardiopulmonary support & cooldown. Just as future
technology may reverse "death " in whole persons, future technology
should also be able to reverse much of what passes for irreversible
death of cells. Certainly we should expect reversibility from the early
stages of apoptosis. Cell death by necrosis should be of much more concern than
apoptosis in cryonics.
The most damaging effect of ischemia within the first hour or two is to the capacity
for cerebral blood flow [BRAIN RESEARCH 81:59-74 (1974)]. Lactic acidosis causes
endothelial cells to swell [ACTA NEUROPATHOLOGIA 60:232-240 (1983)]. Blood cells
stiffen & agglutinate. The longer the ischemia, the worse is the reperfusion injury to blood
vessels due to
free-radicals
& hemorrhage — and the greater the chance of "no reflow" (impeded circulation).
Without circulation there can be no cardiopulmonary support or cryoprotectant perfusion.
By using a cocktail of agents Mike Darwin and Dr. Steve Harris of
Critical Care Research extended the period dogs can tolerate warm
(room-temperature) ischemia to 17 minutes. A cocktail of such agents
reportedly could never pass FDA approval for stroke therapy or cardiac
arrest treatment, hence it did not receive widespread interest or
application in conventional medicine. Dogs have a higher heart rate
and metabolic rate than do humans. The ischemic tolerance for humans
is estimated to be as high as 20 minutes [CRITICAL CARE MEDICINE
16(10):923-941 (1988)].
Under ideal circumstances, however, a cryonics patient experiences
little room-temperature ischemia. If cardiopulmonary support and
cooling are begun immediately ischemia can be minimized. Under
non-ideal circumstances room-temperature ischemia is often
considerably more than 17 minutes.
It is commonly noted that metabolic rate is halved for every 10ºC
drop in temperature. But reducing temperature has a protective effect which
exceeds reduction of metabolism, due to reduction of lipid peroxidation. Experiments
on gerbils indicate that a drop in temperature from 37ºC to 31ºC nearly
triples the amount of time that neurons can tolerate
ischemia [CRITICAL CARE MEDICINE 31(1):255-260 (2003)]. Dogs
cooled to 20ºC can withstand 60 minutes of ischemia and can
withstand 120 minutes of ischemia at 10ºC [CRITICAL CARE
MEDICINE 31(5):1523-1531 (2003)]. Temperatures below 15ºC considerably
reduce ischemic oxidative stress in mice [FREE RADICAL & BIOLOGY
AND MEDICINE; Khandoga,A; 35(8):901-909 (2003)].
A temperature reduction from 37ºC to 26ºC completely inhibited
potassium-induced neurotransmitter release from rat astrocytes
[JOURNAL OF CEREBRAL BLOOD FLOW AND METABOLISM 15:409-416 (1995)].
Marked increases in nitric oxide end-products caused by glutamate infusion in rats
were completely eliminated by reducing temperature from 37ºC to 32ºC [JOURNAL
OF NEUROTRAUMA 20(11):1179-1187 (2003)]. Rats reperfused after a
15-minute ischemic period had over 3 times as many hydroxyl radicals one
hour later than rats subject to ischemia, but not reperfused. But rats
reperfused at 30ºC rather than 36ºC had half as many hydroxyl radicals as
the 36ºC reperfusion rats [JOURNAL OF CEREBRAL BLOOD FLOW AND METABOLISM
16:100-106 (1996)]. The protective effects of hypothermia against ischemic damage
are very nonlinear. Nonetheless, more than a day or two of cold ischemia (4ºC)
greatly reduces survival of kidneys held in organ preservation
solution [TRANSPLANTATION PROCEEDINGS 30:4294-4296 (1998)].
If a cryonics patient is given immediate cardiopulmonary
support, ischemia can be greatly reduced, if not eliminated. Normal
physiologic cerebral blood flow is about 50mL per 100 grams of
brain tissue per minute. Good cardiopulmonary support can maintain
cerebral blood flow not much higher than 15mL (and usually lower),
but only with the assistance of epinephrine [CIRCULATION 69(4):822-835 (1984)].
This is critically close to the 10mL associated with the beginning of
irreversible cell damage if such a flow rate is maintained for an
extended period [JOURNAL OF NEUROSURGERY 77:169-184
(1992)]. Active compression-decompression and interposed abdominal
compression can improve CPR perfusion
considerably [CIRCULATION;100(21):2146-2152 (1999)]
— as can mechanical devices (see below).
With effective cooling the flow provided even with moderately-effective CPR may
be adequate to maintain brain structure. Newton's law of cooling dictates that
temperature drop is most rapid upon initial application of
cooling. And there is a natural drop in brain temperature associated
with reduced blood flow. Under these circumstances the added
benefit of anti-ischemic agents may not be great. (For further discussion of
cooling rates, see my essay Physical Parameters of Cooling in Cryonics.)
These facts should provide some comfort for those who feel
they cannot afford to supplement the cooling and cardiopulmonary
support of cryonics rescue with expensive anti-ischemic cocktails.
Nonetheless, pretreatment of the patient with aspirin, vitamin E and
other anti-oxidants is an inexpensive means of reducing ischemia
after the heart stops. Such pretreatment may give better antioxidant
tissue levels than infusing them after deanimation. because
adenosine
inhibits glutamate release, coffee & tea consumption immediately
prior to deanimation is contraindicated.
High levels of PARP−1 due to high levels of
DNA damage can thus reduce the NAD+ needed for ATP synthesis, leading
to ATP depletion and cell death by necrosis. Or PARP−1 may induce apoptosis by p53
stabilization and/or by translocation of Apoptosis-Inducing Factor (AIF) to the
nucleus [EXPERIMENTAL HEMATOLOGY 31:446-454 (2003)]. PARP−1 inhibitors have been
proposed to protect neurons from excitotoxicity and ischemic damage.
Zinc (Zn2+)
contributes significantly to neuron death in ischemia, but pre-treatment with
EDTA 30 minutes
prior to the ischemic event robustly protects
neurons [THE JOURNAL OF NEUROSCIENCE; Calderone,A; 24(44):9903-9913 (2004)].
Iron and copper can
contribute significantly to free radical damage in ischemia, particularly iron in
cold ischemia because cold ischemia releases iron within cells. Endothelial cells
are significantly more damaged by reperfusion following cold ischemia than following
warm ischemia [TRANSPLANTATION PROCEEDINGS; de Groot,H; 39(2):481-484 (2007)].
The metal chelator deferoxamine has shown signifant benefit against iron-catalyzed
ischemic damage, but deferoxamine does not chelate
copper [JOURNAL OF EXPERIMENTAL BIOLOGY; Warner,DS; 207(18):3221-3231 (2004)].
Other iron chelators have also been shown to be protective [FREE RADICAL BIOLOGY &
MEDICINE; Rauen,U; 37(9):1369-1383 (2004)]. Insofar as blood cells (leukocytes and erythrocytes)
are sources of reperfusion injury damage (cytokines, free radicals and other toxins),
removal of blood cells prior to cold ischemia (shipment of a cryonics patient on ice)
can considerably reduce reperfusion injury (associated with cryoprotective
perfusion) [STROKE; Ding,Y; 33(10):2492-2498 (2002)].
At least two studies have shown that
deprenyl could be of value
in reducing ischemic damage in the brain. A study [STROKE 26:1883-1887 (1995)] involving 14
days of deprenyl on rats and 20 minutes of hypoxia/ischemia
showed reduction of area of damage of 75% in the forebrain and about
20% in the cortex. For the hippocampus, 30-38% of the area was
damaged in controls, but no damage was seen in the depenyl-treated rats.
A similar study on gerbils [JOURNAL OF NEURAL TRANSMISSION 107:779-789 (2000)]
showed reduced damage to the CA1 area of the hippocampus for deprenyl given more than a week before,
immediately after and more than a week after ischemia due to vessel
occlusion. Cell cultures exposed to peroxynitrite have been protected
from apoptotic DNA damage by deprenyl [MECHANISMS OF AGING
AND DEVELOPMENT 111:189-200 (1999)].
Minocycline can reduce inflammation, edema and damage to the blood-brain barrier,
especially when tissue plasminogen activator (tPA) is being used. Activation of MMP−9
by tPA can be countered by the use of
hypothermia [STROKE; Horstmann,S; 34(9):2165-2170 (2003)]. Although opening the
blood-brain barrier is valuable in stroke treatment it may or may not be valuable in
cryonics insofar as opening the blood-brain barrier can assist in getting
cryoprotectants into the brain. (See the earlier sections on
reperfusion injury and
stroke therapy.)
Epinephrine
has commonly been used to maintain blood pressure and
supplement CPR by maintaining blood pressure, although
vasopressin may also be used [CRITICAL CARE MEDICINE
30(supplement 4):S157-S161 (2002)]. Epinephrine, heparin (anti-coagulant),
tPA and even cardiopulmonary support could be counterproductive
for a cryonics patient who has a hemorrhagic
stroke [STROKE; Steiner,T; 37(1):256-262 (2006)].
In hospitals, epinephrine is usually standard for
ACLS
(Advanced Cardiac Life Support). ACLS invariably uses manual
CPR, despite the better blood delivery from mechanical devices. Mechanical
devices are superior to manual CPR because (1) manual CPR
quickly becomes less efficient because it is
much more tiring and (2) manual CPR cannot
deliver as much blood volume in the best of cases because a mechanical
device can deliver a faster high-impulse square-wave compression.
Pronouncement of death may occur soon after the heart stops.
In a Do-Not-Resuscitate (DNR) situation rapid application of CPR could
cause the legally dead person to regain consciousness. It is
unlikely that the heart could restart in an adult — especially if ischemia has
elevated extracellular & plasma potassium levels. The heart rarely
restarts without electonic defibrillation except in young children.
Regaining of consciousness by a cryonics patient would provide
reassurance of the effectiveness of the cardiopulmonary support, but
it would be traumatic for all concerned — and a "political " disaster.
Barbiturates would be an effective means of maintaining
unconsciousness, but as a narcotic its use can be both a political
& legal hazard. Fortunately,
propofol is not a controlled substance
and can keep the patient unconscious. Fortuitously, propofol has also been
shown to inhibit the neural cell apoptosis that can occur as a consequence of
ischemia/reperfusion
injury [THE JOURNAL OF NEUROSCIENCE; Polster,BM; 23(7):2735-2743
(2003)]. Propofol inhibits the opening of the
Mitochondrial
Permeability Transition
Pore (MPTP) [CARDIOVASCULAR RESEARCH;
Javadov,SA; 45(2):360-369 (2000)]. If a funeral director, medical
professional or other person can administer heparin, he or she should
also be able to administer epinephrine, propofol, a thrombolytic,
antioxidants and other agents to combat acidosis.
(For details on more advanced post-mortem changes, see
Postmortem Changes or
Chemistry of Decomposition.)
(For a more in-depth review of cryonics medications,
see Future Directions in Human Cryopreservation Combinational Pharmacotherapy.)
For the terminal cryonics patient it can be asked, why wait until after declaration
of legal death before using
antioxidants or other agents that can reduce
ischemic damage? Higher blood and tissue levels of some antioxidants can be
achieved if administered in the days or weeks before legal death than if
administered after the event. For antioxidants that are legal and safe, a
pre-treatment protocol makes a great deal of sense, although there have been
few controlled studies on such pre-treatment by cryonics researchers or
anyone else. Relevant experiments in the literature generally involve pre-treatment
within one hour prior to induction of ischemia.
Intravenous injection of the alpha-tocopherol form of
Vitamin E (20 mg/kg or
9 mg/pound) 30 minutes prior to ischemia has been shown to significantly
reduce lipid peroxidation and neurological damage [STROKE 14(6):977-982 (1983)].
A better experiment would have included both alpha-tocopherol and gamma-tocopherol
because gamma-tocopherol removes peroxynitrite whereas alpha-tocopherol does
not [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA);
94(7):3217-3222 (1997)].
Vitamin E pretreatment for cryonics patients has the additional advantage of
reducing blood clotting — and does not have the risk of gastric bleeding associated
with aspirin. Many fish oils (especially salmon oil) afford the same benefit, in addition
to reducing the risk of cardiac arrest [MOLECULAR AND CELLULAR BIOCHEMISTRY
116(1-2):19-25 (1992)]. Reduced clotting in a cryonics patient is a great benefit — and is
reason for heparin injection after legal death. For patients undergoing surgery, however,
Vitamin E and fish oils may be prohibited because clotting is desired.
Unlike Vitamin E,
melatonin acts as an antioxidant through endogenous electron
donation, which does not have the same potential for a pro-oxidant side
effect [JOURNAL OF PINEAL RESEARCH 32:135-142 (2002)]. The capacity of melatonin
to scavenge hydroxyl radicals is three orders of magnitude greater than
Vitamin E [JOURNAL OF BIOLOGICAL CHEMISTRY; 274(31):21937-21942 (1999)].
Pretreatment of gerbils with melatonin (10 mg/kg or 4.5 mg/pound) 30 minutes
before reperfusion significantly reduced ischemic brain injury [JOURNAL OF PINEAL
RESEARCH 29:217-227 (2000)]. Similar effects were achieved with rats, but 5mg/kg showed
a greater benefit than a higher or lower dose [JOURNAL OF
PINEAL RESEARCH 34:110-118 (2003)]. Melatonin can also protect against ischemia-reperfusion injury by inhibiting
inducible nitric oxide production, at least partially by means of inhibiting activation of the
pro-inflammatory transcription factor
NF-κB and blockage of NF-κB binding to
DNA [THE FASEB JOURNAL; Gilad,E; 12(9):685-693 (1998)]. Nitric oxide has been shown to exacerbate
apoptosis due to calcium release from the mitochondrial pool and activation of the
Mitochondrial Permeability Transition
Pore (MPTP) [THE FASEB JOURNAL; Horn,TFW; 16(12):1611-1622 (2002)].
Lipoic acid is beneficial in reducing
ischemic-reperfusion injury by direct action as well as by glutathione protection and xanthine
oxidase inhibition [FREE RADICAL BIOLOGY & MEDICINE; Packer, L.; 19(2):227-250 (1995)].
Protection against peroxynitrite damage by lipoic acid is highly dependent upon the target molecule
(some molecules are protected more than others) [JOURNAL OF BIOLOGICAL CHEMISTRY; Rezk,BM; 279(11):9693-9697 (2004)].
Protection of neurons from glutamate excitotoxicity is equally effective by the R-form and
S-form [FREE RADICAL BIOLOGY & MEDICINE; Tirash,O; 26(11/12):1415-1426 (1999)].
CoEnzyme Q10 has been
shown to protect rat endothelial cells from ischemia-reperfusion injury [SURGERY;
Yokoyama,H; 120(2):189-196 (1996)]. Human cardiac arrest patients admitted to a hospital within
6 hours of cardiac arrest given a 250 mg loading dose of CoQ10 showed 68%
survival compared to 30% of controls. Of the survivors, 36% of the CoQ10
group had good neurological outcome, in contrast to 20% of
controls [CIRCULATION; Damian,MS; 110(19):3011-3016 (2004)].
Pretreatment of gerbils with
deprenyl (0.25 mg/kg or 0.11 mg/pound) two weeks
before ischemia reduced damage to neurons in the hippocampus [JOURNAL OF
NEURAL TRANSMISSION 107:779-786 (2000)].
N-acetylcysteine (15 grams) infused
in human myocardial infarction patients over a 24-hour period significantly reduced
ischemic damage [CIRCULATION 92(10):2855-2862 (1995)].
The phytochemical curcumin (which gives curry its yellow color) is a powerful
antioxidant which is several
times more potent than
Vitamin E [THE JOURNAL OF NEUROSCIENCE 21(21):8370-8377 (2001)].
Unlike alpha-tocopherol,
curcumin can scavenge peroxynitrite and inhibit inducible
nitric oxide synthetase [CARCINOGENESIS; Rao,CV; 20(4):641-644 (1999)] — which has the potential
to significantly reduce peroxynitrite damage during reperfusion.
Vitamin C should not be used for ischemia-reperfusion pretreatment. Vitamin C
is an antioxidant in the absence of metal ions, but in the presence of metal ions — which are
released in large quantities from ischemic brain tissue — Vitamin C becomes a powerful
pro-oxidant. (For discussion, see Antioxidant Molecules.)
In sum, a pre-treatment regimen for a terminal cryonics patient weighing 100 kilograms
(220 pounds) should at least contain about 600 mg per day of alpha lipoic acid
(1000 mg per day if racemic rather than R form), 500 mg per day of
CoEnzyme Q10,
and 2,000 IU (mg) per day of mixed tocopherol (equal amounts of alpha and gamma). If the
moment of deanimation (death) can be predicted (as with the removal of life support) then
50 mg of melatonin should be administered 30 minutes before the removal of
life support. Melatonin is quickly metabolized (not stored in tissues) so its value for
extended pretreatment could be debatable. In favor of its use for pretreatment, however, is
the ischemic injury suffered by terminal patients during the dying process (although if
the antioxidants delay the death, the net damage may be the same in the end). Curcumin use
would also be advised, although there is no suggested dose.
When cardiopulmonary support and cooling are initiated soon after
deanimation the use of anti-ischemic agents are probably of marginal
benefit. Pretreatment with high levels of antioxidants, however,
should be easy to do — and be of benefit. Appropriate dosage levels
is guesswork. But it does seem that for antioxidants which have few side effects,
a terminal cryonics patient would benefit by taking dosages which are several
times what would be considered normal for a person taking supplements.
Conditions for cryopreservation are never optimal and so-called substandard treatment
should not be dismissed as being "not worth the effort ". Personal
identity may well survive considerable ischemic damage. Less damage
is better, but not at unlimited cost. Cost/benefit calculations are
difficult to make when benefit is so difficult to quantify. The highest
priority should be to ensure that death does not strike at times & places
that leave one completely unprepared to begin timely cooldown &
cardiopulmonary support.
IV. STROKE THERAPY
V. HIBERNATION AND ESTIVATION
VI. ORGAN TRANSPLANTATION SOLUTION
Adenosine ATP, ADP and AMP
![[ Adenosine ]](./adenosine.gif)
![[ ATP, ADP and AMP ]](./ATP.gif)
VII. BRAIN DAMAGE DUE TO ISCHEMIA/REPERFUSION
VIII. PREVENTING ISCHEMIC/REPERFUSION INJURY IN CRYONICS
IX. PRE-TREATMENT FOR CRYONICS PATIENTS
X. CONCLUSIONS
![[GO TO BEN BEST'S HOME PAGE]](../homeback.gif) HOME PAGE
HOME PAGE